

Designing a medical device that performs beautifully isn’t enough. It also has to meet the world’s toughest regulatory and safety standards.
In 2026, compliance is more than just a checkbox, but, rather, a competitive advantage. Teams that integrate regulation and risk thinking into design early reach the market faster, with fewer redesigns and stronger trust from clinicians and regulators alike.
This article explains the key regulatory frameworks, how risk management fits into every phase of design, and what practical steps keep your project audit ready.
Understanding Device Classification & Regulatory Pathways
Every regulatory journey begins with one simple question:
“What kind of device are we building, and how risky is it?”
The U.S. FDA, the European MDR, and other authorities all classify devices based on risk to the patient.
That classification determines how much evidence, documentation, and testing you’ll need before you can sell.
| Class | Risk Level | Examples | Typical Pathway (U.S.) |
| Class I | Low | Bandages, tongue depressors | General controls |
| Class II | Moderate | Blood pressure monitors, infusion pumps | 510(k) premarket notification |
| Class III | High | Pacemakers, heart valves | Premarket Approval (PMA) |
✔ Pro Tip: Define the device’s intended use and indications for use early.
That wording drives your classification, and therefore your documentation burden, test requirements, and time to approval.
Quality Management Systems (QMS): The Compliance Backbone
A solid QMS is the framework that keeps your design, manufacturing, and post market activities aligned with regulation.
The gold standard is ISO 13485:2016, recognized globally and closely harmonized with the FDA’s Quality System Regulation (21 CFR 820).
What a QMS Should Include:
- Document control and training systems
- Supplier qualification and change control
- Risk management integration (per ISO 14971)
- Design control procedures (inputs, outputs, verification, validation)
- CAPA (Corrective and Preventive Action)
- Post market feedback and complaint handling
✔ Pro tip: Don’t reinvent the wheel. Build your QMS around your design phases.
That ensures compliance feels like part of engineering, not a separate bureaucracy.
Embedding Risk Management in Every Design Phase
Risk management isn’t a single document. It’s a living discipline that evolves as your design matures.
The FDA and ISO 14971 both require continuous identification, evaluation, and mitigation of risks across the entire device lifecycle.
The Risk Management Loop:
- Identify hazards: clinical use, mechanical, electrical, software, data security
- Estimate risk: probability × severity
- Control risk: through design changes, alarms, labeling, or instruction
- Verify effectiveness: confirm controls actually reduce risk
- Document residual risk: justify why it’s acceptable
Example:
| Hazard | Risk | Control | Verification |
| Incorrect dosage | Patient harm | Dual sensor redundancy + software lockout | Simulated failure testing |
| Electrical short | Shock | Grounding and insulation | IEC 60601-1 safety testing |
| Misuse | User injury | Clear UI, labeling, training | Human factors validation |
✔ Pro tip: Maintain a single Risk Register linked directly to your Traceability Matrix. Every user need and design control should map to a risk ID.
Global Regulatory Alignment in 2026
Regulatory harmonization continues to accelerate.
Trends to Note:
- FDA QMSR alignment with ISO 13485: reducing duplication for global manufacturers.
- European MDR & IVDR: more stringent clinical evaluation and post-market surveillance.
- Software as a Medical Device (SaMD): new frameworks for AI, cloud, and algorithmic updates.
- Cybersecurity requirements: explicit FDA premarket guidance since 2023.
✔ Pro tip: Plan for multi-market readiness early.
A design control system built to FDA + ISO standards will also serve Europe, Canada, and Australia with minimal adjustment.
Integration: QMS + Design Controls + Risk
One of the most common pitfalls is treating QMS, risk, and design as separate silos.
In reality, the most successful device teams operate them as one integrated system.
The “Unified Compliance Model”
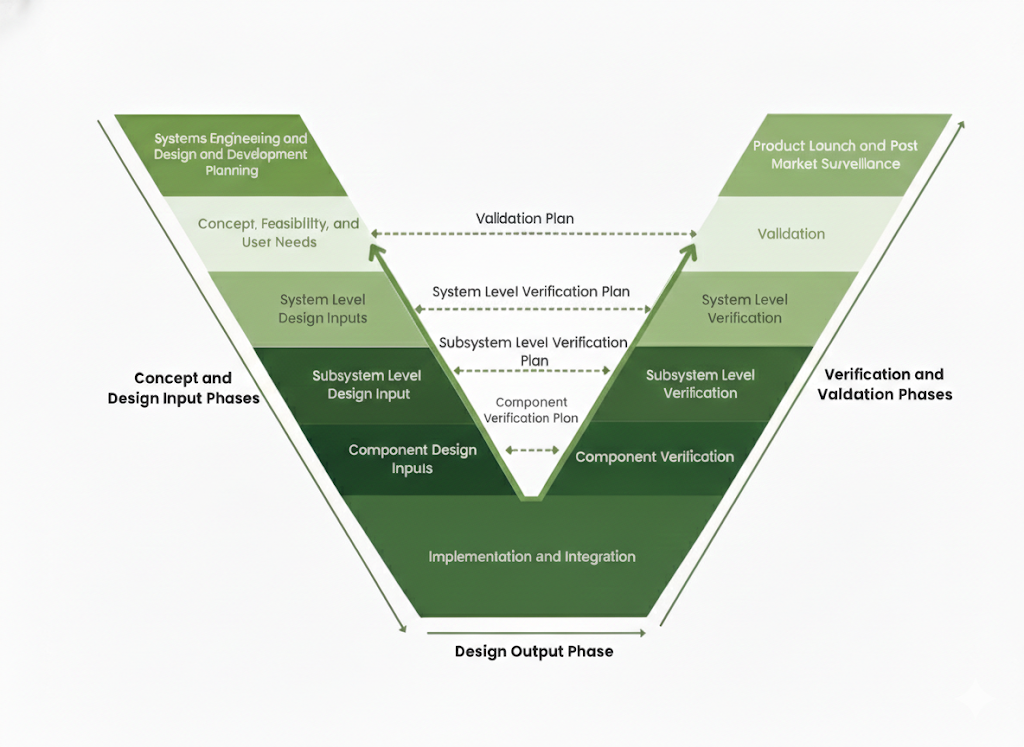
The V-model illustrates how design definition activities on the left map directly to verification and validation activities on the right. Design inputs inform verification planning, user needs drive validation strategy, and risk management shapes both design decisions and verification rigor.
When you synchronize these functions:
- Traceability is clear for every audit.
- Reviews become faster and more factual.
- V&V documentation structure naturally complements design work.
Documentation That Matters
Regulators don’t just want proof you built it right. They want proof you designed it right.
Keep these two files complete and up to date:
| Document | Purpose |
| Design History File (DHF) | Evidence your design followed defined controls (the “how”) |
| Device Master Record (DMR) | The complete recipe for production (the “what”) |
✔ Pro tip: Think of your DHF as your project diary where every decision, test, and change is documented.
Practical Compliance Tips
- Involve regulatory experts at the concept stage.
- Perform internal design reviews at each phase gate.
- Align your test plans with design inputs early.
- Maintain an accessible DHF that requires no last minute document chases.Train your team regularly on QMS updates.
Summary: Compliance is Strategy
Regulatory and risk management are not roadblocks, rather, they’re how you earn trust, accelerate approval, and prevent recalls.
By embedding compliance into your design DNA, you build safer devices, stronger brands, and lasting credibility.
Compliance Review Consultation
Get a free 30 minute consultation to review your regulatory strategy and risk framework.
- Identify classification & documentation gaps
- Map QMS and risk workflows to ISO 13485 and FDA expectations
- Receive practical, audit ready recommendations
Email: sdonnigan@a65consulting.com
Or schedule your compliance readiness review online
References
- U.S. Food and Drug Administration (FDA).
Overview of Medical Device Regulation.
https://www.fda.gov/medical-devices/device-advice-comprehensive-regulatory-assistance/overview-device-regulation - U.S. Food and Drug Administration (FDA).
Classify Your Medical Device.
https://www.fda.gov/medical-devices/overview-device-regulation/classify-your-medical-device - U.S. Food and Drug Administration (FDA).
Quality System Regulation (21 CFR Part 820).
https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-820 - U.S. Food and Drug Administration (FDA).
Design Control Guidance for Medical Device Manufacturers.
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/design-control-guidance-medical-device-manufacturers - International Organization for Standardization (ISO).
ISO 13485:2016 — Medical devices: Quality management systems — Requirements for regulatory purposes.
https://www.iso.org/standard/59752.html - International Organization for Standardization (ISO).
ISO 14971:2019 — Medical devices: Application of risk management to medical devices.
https://www.iso.org/standard/72704.html - U.S. Food and Drug Administration (FDA).
Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions.
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/cybersecurity-medical-devices-quality-system-considerations-and-content-premarket